Neurologia molecular: És sabut que els pèptids beta-amiloide (Aβ) juguen un paper important en la patogènesi de la malaltia d’Alzheimer. Aquests pèptids deriven d’una proteïna precursora, designada com a APP. Conèixer quin és el paper fisiològic de l’APP pot ésser rellevant e la caracterització dels mecanismes molecular de les malalties neurodegeneratives. Amb aquesta intenció Mel B. Feany, professora de patologia de la Harvard Medical School, ha coordinat una anàlisi integrativa que apareixia ahir en forma d’article a Nature Communications, amb Vanitha Nithianandam com a primera autora. En l’article expliquen han recuperat Appl, la proteïna ortòloga de l’APP en Drosophila melanogaster en un rastreig de mutants neurodegeneratius d’aquesta mosca de la fruita. Investiguen la funció d’Appl en el cervell de mosques d’edat avançada mutants per al gen Appl a través d’estudis transcripcionals i proteòmics de cèl·lules aïllades. Nithianandam et al. troben que Appl té un rol en el control de múltiples processos cel·lulars, com ara la traducció d’ARNm a proteïna, la funció mitocondrial, el metabolisme d’àcids nucleics, el metabolisme lipídic, les vies de senyalització o la proteòstasi (l’equilibri entre la síntesi i degradació de proteïnes). L’Appl regularia l’autofàgia a través de la via de senyalització del TGFβ. Això no és quelcom exclusiu del cervell de Drosophila, tal com constaten Nithianandam et al. en models genètics de ratolins, de cèl·lules mare induïdes humanes (iPSC) i de tauopaties in vivo. Tot plegat suggereix que APP té un rol en la regulació de la proteòstasi en el cervell d’animals ben diferents.
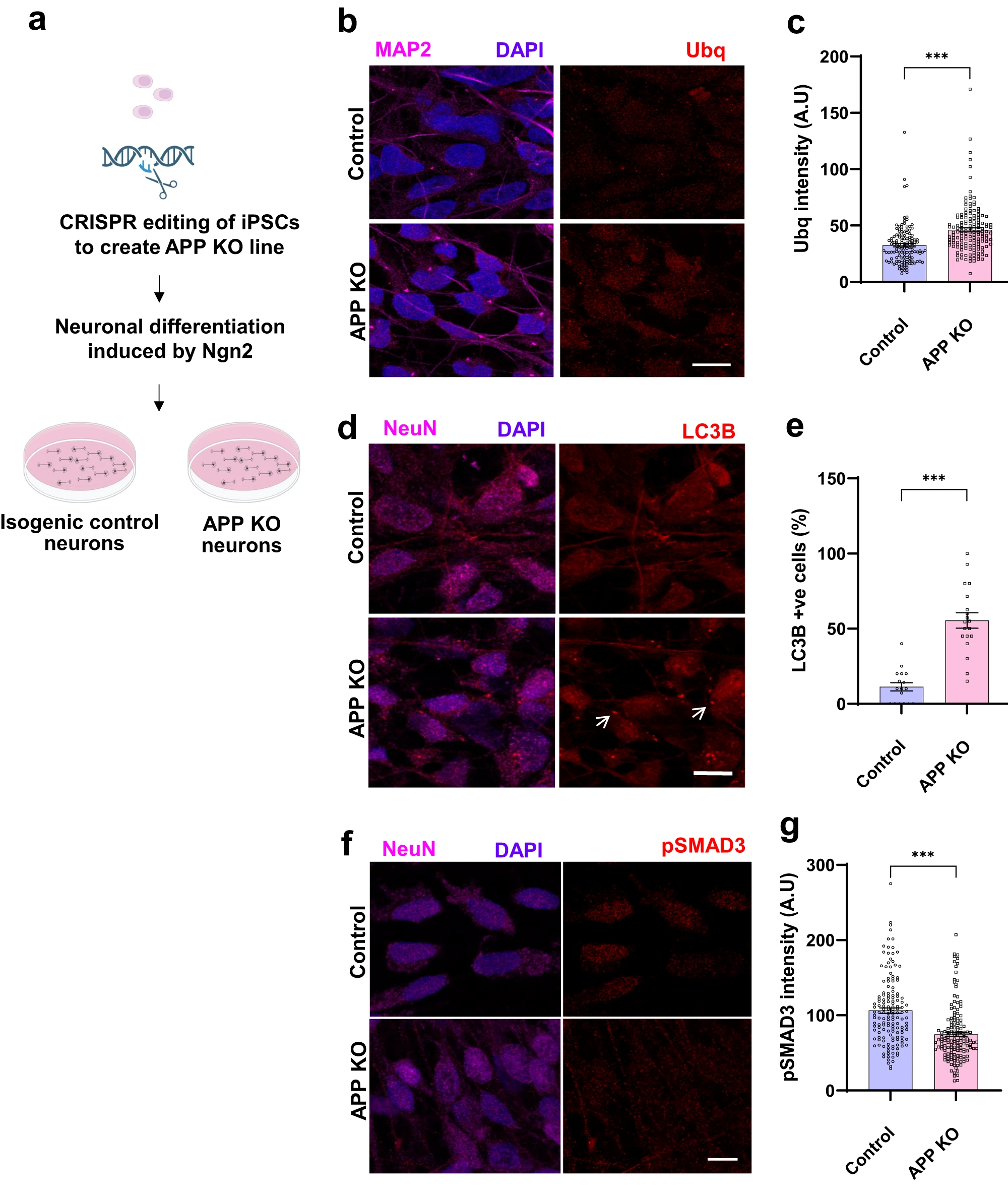
La creació d’una línia de cèl·lules humanes de totipotència induïda a les que s’ha eliminat el gen APP a través de la tecnologia CRISPR mostra el rol de la proteïna APP en la proteòstasi quan aquesta línia és diferenciada en neurona
La proteïna precursora d’amiloide
Nithianandam i Feany han dissenyat aquest estudi. Els experiments foren realitzats per Nithianandam, Feany, Hassan Bukhari (Harvard), Matthew J. Leventhal (MIT), Rachel A. Battaglia (Harvard), Xianjun Dong (Harvard), Ernest Fraenkel (MIT) realitzaren experiments i/o analitzaren dades. La recerca s’ha finançat per la Michael J. Fox Foundation for Parkinson’s Research i la Ellison Medical Foundation. L’article fou tramès a la revista el 18 d’abril del 2023. Després d’un procés de revisió, l’article fou acceptat el 23 d’octubre i publicat el 3 de novembre.
La proteïna precursora amiloide (APP) és una proteïna transmembrana que consta d’un gran domini extracel·lular, un únic domini transmembrana i un petit domini intracel·lular. L’acció de la proteasa β-secretasa sobre el domini extracel·lular de l’APP i l’acció del complex ɣ-secretasa sobre el domini transmembrana resulta en la producció de pèptids, els quals s’agreguen en plaques extracel·lulars. Aquestes plaques amiloides són un signe requerit per al diagnòstic de la malaltia d’Alzheimer. La formació d’aquests pèptids intervindria en la patogènesi de l’Alzheimer. Hom ha descrit mutacions familiars en APP en formes de malaltia d’Alzheimer d’herència autosòmica i d’alta penetrància. Aquestes mutacions en concentren en els aminoàcids propers als llocs de tall de l’APP per les secretases. Altres formes familiars d’Alzheimer són provocades per mutacions d’alta penetrància en les proteïnes per aquesta raó denominades presenilines, que participen en el complex ɣ-secretasa. Totes aquestes mutacions tendeixen a afavorir la formació de pèptids amiloides de 42-43 aminoàcids (Aβ42-43), en detriment de la forma de 40 aminoàcids (Aβ40): els primers afavoreixen una major acumulació de plaques amiloides (són més amiloidogènics). En un sentit contrari una variant rara d’APP que afecta un lloc proper al tall de la β-secretasa redueix el risc de patir declivi cognitiu amb l’edat.
Bo i que hi ha un gran coneixement sobre el paper patològic dels pèptids beta-amiloides (Aβ) en la malaltia d’Alzheimer, no hi ha pas tant sobre el rol fisiològic de la proteïna precursora, l’APP. En vertebrats, a més de l’APP, hi ha dues proteïnes semblants més, APLP1 i APLP2. Ratolins knockout per al gen APP són viables i amb pocs signes d’anormalitat. En canvi, ratolins amb doble knockout per als gens APP i APLP2 o amb triple per a APP, APLP1 i APLP2, no ho són (pateixen mortalitat perinatal).
Appl en Drosophila
En la mosca de la fruita (Drosophila melanogaster), hi ha un únic gen de la família APP, Appl. Mosques sense el gen Appl són viables.
Nithianandam et al. realitzaren un triatge genètic en Drosophila melanogaster a la recerca de proteïnes i processos requerits en el manteniment de la viabilitat neuronal amb l’edat. Entre les proteïnes identificades aparegué l’Appl. Seguidament, Nithianandam et al. feren una anàlisi transcriptòmica i proteòmica de mosques mutants per a l’Appl que indicava la importància d’aquesta proteïna en el control de la proteòstasi.
El triatge aplicat per Nithianandam et al. utilitza ARN d’interferència (RNAi) transgènic per induir un knock down de gens individuals. Com que el RNAi utilitzat deriva d’ARN missatger de neurones (pan-neuronal), inactivarà gens que s’expressen en aquest tipus cel·lular. Les mosques obtingudes en aquest triatge foren sacrificades a 30 dies d’edat: es preparaven seccions histològiques del cap i s’estudiava el nivell de neurodegeneració del cervell. En total s’analitzaren 6.258 gens diferents, dels quals el noquejament de 307 conduïa a una major neurodegeneració. Entre aquests 307 gens hi havia el gen Appl.
A través d’una deleció sintètica de la porció central del gen Appl, Nithianandam et al. crearen l’al·lel defectiu Appld. Les mosques amb aquest al·lel no expressen la proteïna Appl. En mosques de més de 30 dies amb aquesta mutació a les que s’havia introduït un reportador transgènic de l’activitat caspasa (relacionada amb l’apoptosi o mort cel·lular programada), Nithianandam et al. detectaven un augment d’aquesta activitat en neurones. Paral·lelament s’observa un augment del nombre de vacuoles neurodegeneratives en el neuropil i el còrtex del cervell d’aquestes mosques.
Nithianandam et al. utilitzen la seqüenciació unicel·lular d’ARN (scRNA) per identificar ARNm que siguin regulats per Appl en diferents tipus cel·lulars. Comparen així cervells desagregats de mosques control i de mosques Appld, de manera que abasten neurones, glia, hemòcits, cèl·lules del cos gras, cèl·lules còniques, etc. Així identifiquen 720 gens regulats per l’Appl a l’alça i 717 gens regulats a la baixa. Entre els primers hi ha gens mitocondrials. En els segons hi ha gens implicats en la síntesi proteica. Appl regularia la transcripció de gens implicats en la traducció d’ARNm, desenvolupament i funcionament d’axons i dendrites, funció sinàptica, adhesió cel·lular o memòria a llarg termini.
Com que Appl s’expressa principalment en neurones, Nithianandam et al. examinen especialment els resultats d’aquest tipus cel·lular. Així troben que Appl atenua la senyalització TGFβ/BMP.
Amb espectrometria de masses, Nithianandam et al. realitzen una anàlisi proteòmica i ubiqüitinòmica en caps de mosques control i Appld. La identificació de les proteïnes ubiqüitinilades (destinades a degradació pel proteasoma) es fa amb anticossos antidiglicina. Amb l’anàlisi proteòmica s’identifiquen 6.364 proteïnes, de les quals 545 són regulades a l’alça per Appl i 679 regulades a la baixa. Amb l’anàlisi ubiqüitinòmica s’identifiquen 41 proteïnes amb una major senyalització per Appl i 128 amb una de menor. Tot plegat indica que l’eliminació de l’Appl produeix una alteració en tres processos proteostàtics: la síntesi proteica, el plegament de proteïnes i la degradació de proteïnes.
La immunotinció de talls histològics de cervell de mosques amb anticossos anti-ubiqüitina indica que en les mosques Appld hi ha un augment d’agregats ubiqüitinilats en la retina i en el cervell. La introducció en aquestes mosques de l’expressió de la isoforma humana d’APP de 695 aminoàcids redueix aquests agregats. En canvi, quan s’interfereix (amb RNAi) l’expressió de components de la via de senyalització de TGFβ hi ha un augments dels agregats.
Amb anticossos anti-Atg8a, Nithianandam et al. segueixen la formació d’autofagosomes en cervell de mosques, i hi troben un augment en les mosques Appld. Amb anticossos anti-p62 també detecten un augment de la presència del sequestosoma 1 en els agregats retinals de Appld. Aquestes acumulacions es poden revertir amb l’expressió neuronal de la forma secretada d’Appl (Appl-s).
La deficiència d’APP en neurones de ratolí
En el model de doble deleció d’APP i d’APLP2 de ratolí (N-dCKO), Nithianandam et al. estudien cervells d’animals de 18 mesos d’edat, i els comparen amb controls. Les neurones corticals mostren un augment dels nivells d’ubiqüitina, LC3B i GABARAP, tots ells vinculats a l’autofàgia. També hi troben una reducció de la fosforilació de SMAD3, component de la via de senyalització de TGFβ.
La deficiència d’APP en neurones humanes
Nithianandam et al. utilitzen cèl·lules humanes a les que s’ha induït pluripotència (iPSC). Amb una tècnica CRISPR eliminen el gen APP en les iPSC, i després les diferencien en neurones amb l’expressió de la neurogenina2, alhora que les marquen amb la proteïna fluorescent verda (GFP). Com a controls utilitzen neurones isogèniques derivades d’iPSC que conserven el gen APP. Amb un Western blot s’asseguren que les neurones APP- no expressin la proteïna APP.
Les neurones humanes APP-, comparades amb els controls, mostren nivells més elevats d’ubiqüitina citoplasmàtica. També augmenten els nivells de marcadors autofàgics com LC3B i GABARAP. En el cas de GABARAP hi ha un augment de la ratio de GABARAP-I (forma citosòlica) respecte de GABARAP-II (la forma conjugada amb fosfatidiletanolamina que s’uneix a membranes cel·lulars). També hi ha una disminució de la forma fosforilada de SMAD3 en el nucli cel·lular.
APP i tauopatia
A més de les plaques d’Aβ, el diagnòstic post-mortem de la malaltia d’Alzheimer requereix la identificació de masses neurofibril·lars intracel·lulars de proteïna tau. La proteïna tau en una proteïna d’unió a microtúbuls.
La inducció de l’expressió d’una forma mutant de la proteïna tau humana en tot el sistema nerviós de Drosophila ha estat utilitzada pel grup de recerca de Feany com un model in vivo de tauopatia.
Aquest model de tauopatia aplicat a mosques Appld mostra un impacte addicional en la funció locomotriu. La neurotoxicitat tau també es dispara en forma d’una major apoptosi i formació de vacuoles en el cervell. L’esperança de vida d’aquestes mosques es redueix respecte dels controls.
Aquesta funció d’APP o Appl en la preservació de la proteòstasi fa qüestionar a Nithianandam et al. la viabilitat de promoure la depleció d’APP com a opció terapèutica en la malaltia d’Alzheimer. Aquesta depleció pot ajudar a reduir l’acumulació de pèptids amiloides, però també faria perdre la contribució fisiològica de l’APP.
Lligams:
- Integrative analysis reveals a conserved role for the amyloid precursor protein in proteostasis during aging. Vanitha Nithianandam, Hassan Bukhari, Matthew J. Leventhal, Rachel A. Battaglia, Xianjun Dong, Ernest Fraenkel, Mel B. Feany. Nature Communications 14: 7034 (2023).