Biologia molecular: L'Acadèmia Reial Sueca de Ciències ha anunciat la concessió del Premi Nobel de Química a David Baker "pel disseny computacional de proteïnes" i a Demis Hassabis i John M. Jumper "per la predicció de l'estructura de proteïnes". Baker rebrà la meitat del guardó, és a dir 5,5 milions de kronor sueques. Hassabis i Jumper rebran conjuntament l’altra meitat.
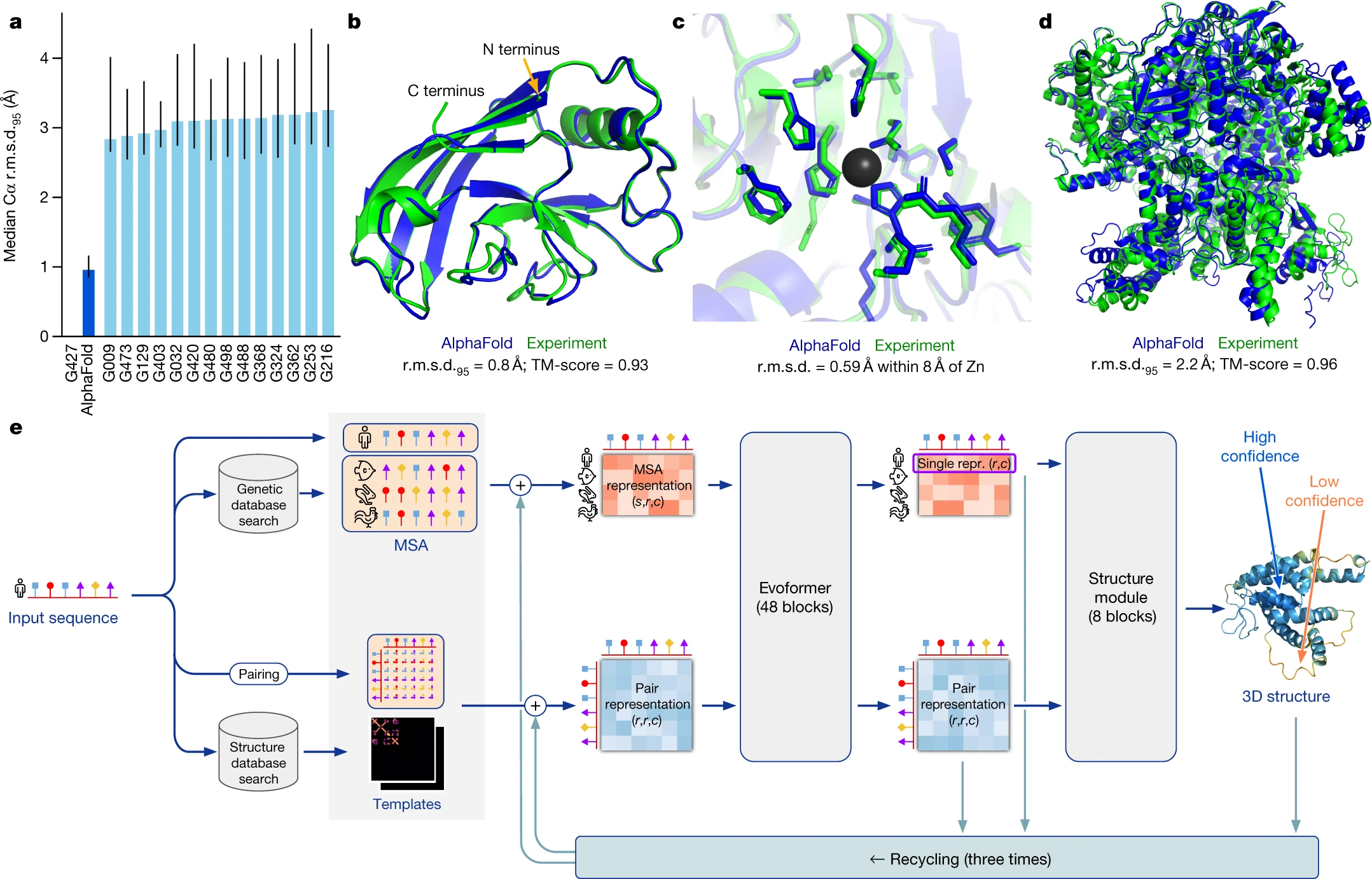
Esquema de funcionament d’AlphaFold2, desenvolupat per Demis Hassabis i John M. Jumper, eina capaç de deduir l’estructura tridimensional d’una proteïna a partir de la seva seqüència aminoacídica.
David Baker

David Baker (*Seattle, 6.10.1962) va nàixer al si de la família jueva formada pel físic Marshall Baker i la geofísica Marcia Bourgin Baker. Es graduà a la Havard University el 1984. Realitzà la tesi doctoral a la University of California, Berkeley, en el laboratori de Randy Schekman (*1948) sobre el transport i tràfic de proteïnes en el llevat, que defensà reeixidament en el 1989. Entre el 1989 i el 1993 fou investigador postdoctoral en el laboratori de biofísica de David Agard, de la UC, San Francisco. El 1993 començà a treballar en el Department of Biochemistry de la University of Washington School of Medicine. Actualment és professor de bioquímica a la University of Washington Seattle, director de l’Institute for Protein Design i investigador del Howard Hughes Medical Institute (des del 2000). És el marit de la bioquímica Hannele Ruohola-Baker (*1959), amb la que tingué dos fills.
Demis Hassabis

Demis Hassabis (*Londres, 27.7.1976) és fill d’un matrimoni del nord de Londres format per un greco-xipriota i una singapuresa. A quatre anys ja destacava com un prodigi dels escacs, guanyant premis per a l’equip infantil d’Anglaterra. Entre el 1988 i el 1990 estudià al Queen Elizabeth’s School, Barnet. Més endavant fou educat a casa pels pares. Amb els premis dels escacs, comprà un ZX Spectrum 48K i estudià pel seu compte programació. Després fins a 16 anys estudià al Christ’s College, Finchley. Sol·licità llavors l’ingrés a la Cambridge University, la qual li recomanà d’esperar-se un any, que aprofità per treballar en disseny de videojocs a Bullfrog Productions. Allà intervingué en els jocs Syndicate i Theme Park (1994), col·laborant amb Peter Molyneux (*1959). Passà després al Queen’s College, Cambridge, on es graduà en informàtica (1997). Llavors entrà a treballar a Lionhead Studios, fundada per Molyneux. El 1998 fundà ell mateix Elixir Studios, que va vendre el 2005. Sota la supervisió d’Eleanor Maguire (*1970) inicià una tesi doctoral en neurociència cognitiva que defensà reeixidament en el 2009. Treballa després en el laboratori de Tomaso Poggio (*1947) al MIT i a la Harvard University. Més endavant fou investigador postdoctoral sota Peter Dayan al Gatsby Computational Neuroscience Unit del UCL. Investigà en el camps de la imaginació, la memòria i l’amnèsia. Identificà la construcció d’escena com a element central del sistema de memòria episòdica (Hassabis & Maguire, 2007). En el 2010 co-fundà DeepMind com a start-up d’intel·ligència artificial, que en el 2014 fou adquirida per Google. Actualment treballa a Google DeepMind.
John M. Jumper

John Michael Jumper (*Little Rock, 1985) es graduà en física i matemàtica a la Vanderbilt University. Després passà a la University of Cambridge on realitza una tesina de màster sobre física teòrica de matèria condensada. Es doctorà en el 2017 a la University of Chicago amb una tesi sobre l’aplicació de l’aprenentatge mecànica en la simulació del plegament i dinàmica de proteïnes, sota la supervisió conjunta de Tobin R. Sosnick i Karl Freed (*1942). Actualment treballa a Google DeepMind.
Disseny computacional de proteïnes
El disseny computacional de proteïnes permet aconseguir proteïnes amb noves funcions i característiques que hom no troba en les proteïnes naturals. David Baker fou pioner en aquest disseny computacional.
Les proteïnes són les eines químiques fonamentals de la matèria viva. Controlen i catalitzen la pràctica totalitat de les reaccions químiques dels sistemes biològics. Una part de les hormones, moltes substàncies senyalitzadores, tots els anticossos i la majoria dels elements estructurals dels teixits són proteïnes.
Malgrat l’ample ventall de funcions i formes, la base constitutiva de les proteïnes es redueix pràcticament a 20 tipus diferents d’aminoàcids. Una cadena peptídica de n aminoàcids pot presentar 20n combinacions, i la maquinària de síntesi proteica pot generar-les totes.
David Baker havia desenvolupat el programa Rosetta de predicció d’estructures tridimensionals de proteïnes a partir de la seqüència peptídica. Raonà que no pas menys interessant seria predir la seqüència peptídica a partir de l’estructura tridimensional. L’estructura tridimensional informa de la funcionalitat de la proteïna, i amb una Rosetta inversa seria possible inventar noves proteïnes.
Altres grups de recerca treballaven en disseny de proteïnes o en enginyeria de proteïnes. En general, es tractava de projectes de modificació puntual de la seqüència aminoacídica per aconseguir proteïnes adaptades a nous substrats o a noves condicions de temperatura, salinitat, pH, etc. La perspectiva de Baker era diferent: es tractava de crear proteïnes de novo a partir de la funció desitjada.
En el 2003 Baker assolí el disseny d’una nova proteïna sense analogia amb les proteïnes biològiques. El primer pas era definir una nova estructura proteica d’interès. Una vegada definida, s’emprava el programa Rosetta per computar quina seqüència aminoacídica seria capaç de generar tal estructura. Per fer-ho el programa rastrejava les bases de dades de proteïnes, considerava aspectes bioenergètics i proposava una seqüència. Fou així com Baker desenvolupà la proteïna Top7. Rosetta proposà una seqüència aminoacídica. Aplicant-hi el codi genètic invers, el grup de Baker obtingué la seqüència gènica corresponent, i introduí aquest gen artificial en un vector d’expressió bacterià. La proteïna sintetitzada fou cristal·litzada i analitzada per raigs X, obtenint una estructura gairebé exacta a la predefinida. Top7 és una proteïna globular de 93 aminoàcids sense cap analogia a la natura (Kuhlman et al., 2003).
El seu laboratori ha creat des de llavors proteïnes sintètiques amb aplicacions farmacèutiques, vaccinals, en nanomaterials i en nanosensors. En el 2008 dissenyaven enzims retro-aldol (Jiang et al., 2008). En el 2013 obtenien proteïnes de síntesi capaces d’unir-se a la digoxigenina amb alta afinitat i selectivitat (Tinberg et al., 2013). En el 2016 el grup de Baker havia creat nous nanomaterials capaços de vincular espontàniament fins a 120 proteïnes. En el 2017 crearen proteïnes capaces d’unir-se a l’opioide fentanil, que podrien fer-se servir com a detectors ambientals d’aquesta substància (Bick et al., 2017). Rosetta fou millorat amb la introducció de models d’intel·ligència artificial basats en xarxes neurals transformadores (Baek et al., 2021). Així en el 2021 crearen nanopartícules amb proteïnes que imiten el virus de la grip en superfícies: han funcionat com a vaccins en models animals. En el 2022 dissenyaren una proteïna que funciona com a rotor molecular. En el 2024 desenvoluparen proteïnes amb formes geomètriques modulables per variables externes, i per tant que poden funcionar com a sensors.
Predicció de l'estructura de proteïnes
Demis Hassabis i John Jumper aconseguiren resoldre mitjançant una eina d'intel·ligència artificial, AlphaFold, el problema de predir l’estructura tridimensional d'una proteïna a partir de la seqüència de pèptids que la conformen.
Les proteïnes són en darrer polímers d’aminoàcids units per enllaços peptídiques. L’estructura primària d’una proteïna és aquesta seqüència lineal d’aminoàcids. Les cadenes laterals dels aminoàcids, però, determinen encara una estructura secundària. La globalitat de la proteïna, en interacció amb el medi, adopta un plegament que defineix una estructura terciària. Les interaccions entre proteïnes i amb altres grups bioquímics (grups prostètics) defineix una estructura quaternària. La seqüència aminoacídica d’una proteïna ve determinada, en termes generals, per la seqüència nucleotídica del gen corresponent. Un codi genètic, definit per la maquinària formada per ribosomes, ARN de transferència i aminoacil-ARN-transferases, i de caràcter gairebé universal per a tots els organismes, relaciona una i l’altra. L’estructura final de la proteïna, elucidada mitjançant tècniques de cristal·lografia de raigs X des dels anys 1950, però, no respon a un automatisme directe sobre l’estructura primària, com sí va pensar inicialment Christian Anfinsen (1916-1995). Una mateixa proteïna, per exemple, pot adoptar plegaments diferents segons el context. Cyrus Levinthal (1922-1990) calculà en el 1969 que una petita proteïna de 100 aminoàcids pot adoptar 1047 estructures tridimensionals diferents. Si el plegament fos aleatori, el camí per arribar a l’estructura tridimensional correcte trigaria eons, però en la cèl·lula això es fa en qüestió de milisegons. La paradoxa s’explica perquè en els sistemes biològics el plegament de proteïnes es troba altament regulat, i no poques patologies poden explicar-se com a errors de plegament de proteïnes. Durant mig segle romangué obert el problema de si era realment possible predir l’estructura d’una proteïna a partir de la seqüència aminoacídica o bé a partir de la seqüència nucleotídica del seu gen. El desenvolupament de les tècniques de seqüenciació gènica fa que a les bases de dades hom disposi més fàcilment de dades sobre la seqüència gènica que no pas sobre l’estructura final de la proteïna corresponent. En l’actualitat hi ha dades de seqüència nucleotídica de 3.000 milions de gens i dades de seqüència aminoacídica de més de 200 milions de proteïnes, però hom no disposa d’imatges per cristal·lografia de raigs X més que de 200.000. Ara bé, és l’estructura tridimensional la que determina la funció d’una proteïna, la capacitat d’interacció amb altres proteïnes i la capacitat de catalitzar tal o tal altra reacció química.
En el 1994 fou bastí el projecte CASP (Critical Assessment of Protein Structure Prediction). Amb una freqüència biennal els investigadors participants rebien accés a la seqüència aminoacídica de proteïnes de les quals s’havia determinat l’estructura per cristal·lografia de raigs X. Els participants, però, no tenien accés a les dades de raigs X, sinó que havien de predir-les a partir de la seqüència.
No fou fins a CASP13 (2018) que aparegueren els primers avenços significatius. Arribaren de la mà de Hassabis, amb un currículum de mestre d’escacs, expert en neurociència i pioner en intel·ligència artificial. En les edicions prèvies de CASP els millors resultats consistien en una precisió del 40%. L’equip de Hassabis, amb el model d’intel·ligència artificial AlphaFold aconseguí una precisió del 60%.
Un altre de participants de CASP13 fou David Baker. Baker havia desenvolupat el programari Rosetta de predicció d’estructures de proteïna. Des del 1993 havia treballat en experiments de plegament de proteïnes. Rosetta quedà lluny dels resultats d’AlphaFold, però superà la majoria dels altres competidors (Simons et al., 1999).
Els resultats d’AlphaFold del 1998 eren, però, lluny de l’objectiu d’una precisió del 90%. John Jumper s’afegí a l’equip, amb el bagatge d’haver ideat mètodes per a la simulació de dinàmica de proteïnes. Així s’inicià el projecte d’AlphaFold2. L’eina fou entrenada amb les seqüències aminoacídiques de totes les proteïnes de les quals es coneix l’estructura (un parell de centenars de milers). En la fase executiva, AlphaFold2 rep una seqüència aminoacídica problema. El primer pas que fa és alinear-la amb totes les seqüències conegudes a les bases de dades per tal d’identificar-hi els aminoàcids més ben preservats evolutivament. El segon pas és explorar quins aminoàcids de la cadena poden interactuar entre ells en l’estructura tridimensional: per exemple els aminoàcids de carrega iònica negativa interactuen amb els de càrrega positiva; els aminoàcids de cadena lateral hidròfoba interactuen entre ells; etc. Amb un procés interatiu, AlphaFold2 refina el mapa de distàncies entre aminoàcids: aquest procés fa servir les xarxes neurals transformadores proposades per Jumper. L’estructura tridimensional obtinguda és analitzada fins a tres cicles abans de fixar una resposta i calcular-ne la probabilitat.
En el 2020 Hassabis i Jumper presentaren un model d’intel·ligència artificial denominat AlphaFold2 (Senior et al., 2000; Jumper et al., 2021). Aquest model fou capaç de predir l’estructura de gairebé totes les proteïnes llavors presents en les bases de dades, que pujaven a 200 milions. En els darrers quatre anys, AlphaFold2 ha estat emprat per més de dos milions d’usuaris de 190 països diferents. Entre les aplicacions que han trobat aquests usuaris hi ha un millor coneixement dels mecanismes de resistència microbiana a antibiòtics o l’elucidació dels mecanisme que permet alguns enzims de degradar plàstics.
Lligams:
- Pressmeddelande: Nobelpriset i kemi 2024.
- Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker. Science 302: 1364-1368 (2003).
- Improved protein structure prediction using potentials from deep learning. Andrew W. Senior, Richard Evans, John Jumper, James Kirkpatrick, Laurent Sifre, Tim Green, Chongli Qin, Augustin Žídek, Alexander W. R. Nelson, Alex Bridgland, Hugo Penedones, Stig Petersen, Karen Simonyan, Steve Crossan, Pushmeet Kohli, David T. Jones, David Silver, Koray Kavukcuoglu, Demis Hassabis. Nature 577: 706-710 (2020).
- Highly accurate protein structure prediction with AlphaFold. John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, Alex Bridgland, Clemens Meyer, Simon A. A. Kohl, Andrew J. Ballard, Andrew Cowie, Bernardino Romera-Paredes, Stanislav Nikolov, Rishub Jain, Jonas Adler, Trevor Back, Stig Petersen, David Reiman, Ellen Clancy, Michal Zielinski, Martin Steinegger, Michalina Pacholska, Tamas Berghammer, Sebastian Bodenstein, David Silver, Oriol Vinyals, Andrew W. Senior, Koray Kavukcuoglu, Pushmeet Kohli, Demis Hassabis. Nature 596: 583-589 (2021).
- Accurate prediction of protein structures and interactions using a three-track neural network. Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Rie Lee, G.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; Millan, C.; Park, H.; Adams, C.; Glassman, C.R.; DeGiovanni, A.; Pereira, J.H.; Rodrigues, A.V.; van Dijk, A.A.; Ebrecht, A.C.; Opperman, D.J.; Sagmeister, T.; Buhlheller, C.; Pavkov-Keller, T.; Rathinaswamy, M.K.; Dalwadi, U.; Yip, C.K.; Burke, J.E.; Garcia, K.C.; Grishin, N.V.; Adams, P.D.; Read, R.J.; Baker, D. Science 373: 871-876 (2021).
Cap comentari:
Publica un comentari a l'entrada