Hematologia: L’Agència Reguladora de Medicina i Productes d’Atenció de la Salut del Regne Unit (MHRA) anunciava ahir l’autorització de la primera teràpia gènica al món que cerca la guarició de l’anèmia falciforme i de la β-talassèmia dependent de transfusions. Aquestes són dues hemoglobinopaties hereditàries degudes a variants dels gens de la globina que, si bé en heterozigosi confereixen una certa resistència a la malària, en homozigosi condueixen a una malaltia severa. Són les comunitats ètniques d’origen africà i mediterrani les que presenten una major prevalença al Regne Unit d’aquestes condicions. La teràpia aprovada ahir rep el nom de Casgevy i consisteix en cèl·lules mare autòlogues (autotemcel) que han estat modificades genèticament ex vivo (exagamglogen) gràcies a l’eina d’edició genètica CRISPR.
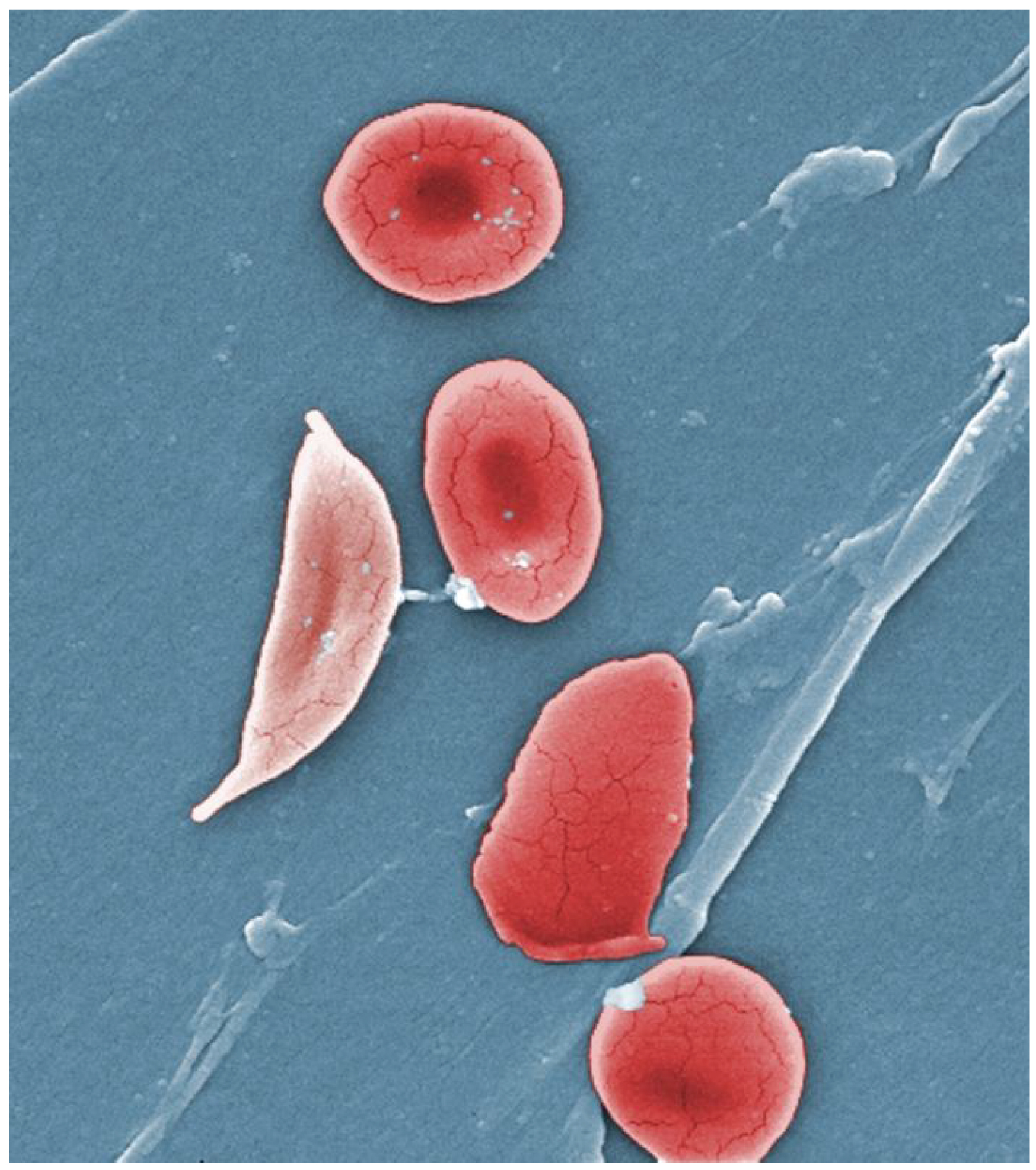
Electromicrografia de transmissió que mostra un eritròcit falciforme al costat d’altres eritròcits
Un nou tractament per a l’anèmia falciforme
La MHRA ha autoritzat aquest nou tractament a pacients majors de 12 anys afectats bé per anèmia falciforme bé per la beta-talassèmia dependent de transfusions sanguínies. La decisió s’ha pres després de la realització d’assaigs sobre la seguretat, qualitat i efectivitat del tractament.
El nou tractament rep el nom de Casgevy, i és la primera medicina aprovada per una agència del medicament que fa ús de l’eina d’edició genètica CRISPR.
Tant l’anèmia falciforme com la β-talassèmia són malalties genètiques degudes a mutacions detrimentals en gens que codifiquen les cadenes proteiques de l’hemoglobina, el pigment de transport d’oxigen que dóna el color característic a la sang. Els glòbuls vermells o eritròcits són el principal component corpuscular de la sang, i funcionen com a autèntics sacs d’hemoglobina. L’hemoglobina de pacients amb anèmia falciforme (hemoglobina S) provoca la deformació dels eritròcits que passen a adquirir una morfologia de falç per comptes de la normalment discoidal. L’anèmia falciforme és sobretot habitual en persones de descendència africana o caribenya i afecta unes 15.000 persones al Regne Unit. La beta-talassèmia ho és principalment en persones de descendència mediterrània o del sud d’Àsia.
Les alteracions moleculars i corpusculars de l’anèmia falciforme es manifesten en atacs de dolor sever, en infeccions greus i perilloses, a més de la pròpia anèmia.
Les alteracions moleculars de la beta-talassèmia es manifesten en anèmia severa, sovint amb la necessitat de rebre transfusions sanguínies cada 3-5 setmanes.
La finalitat de Casgevy és la d’editar el gen defectuós en les cèl·lules mare hematopoiètiques del moll de l’os d’aquests pacients. Així s’aconsegueix que el moll de l’os produeixi eritròcits amb una hemoglobina plenament funcional.
Teràpia gènica i teràpia cel·lular
El tractament Casgevy comporta un primer pas d’extracció de cèl·lules mare hematopoiètiques del propi pacient. Aquestes cèl·lules són tractades en el laboratori amb una tècnica d’edició genètica, i la població cel·lular resultant és transplantada al pacient. És, doncs, un autotransplantament de moll de l’os (o transplantament de cèl·lules hematopoiètiques autòlogues), però amb la particularitat que les poblacions cel·lulars transplantades passen per un procés extracorporal d’edició genètica.
Els assaigs clínics
En l’assaig clínic de Casgevy per a l’anèmia falciforme han participat 45 pacients. D’aquests hom disposa d’un període prou llarg de seguiment per a 29. D’aquests 29, n’hi ha 28 (97%) que no han patit cap atac dolorós en l’any posterior al tractament.
En l’assaig clínic de Casgevy per a la beta-talassèmia dependent de transfusió han participat 54 pacients. Dels 42 pacients dels quals hom disposa de prou temps de seguiment, 39 (93%) no han necessitat de cap transplantament de glòbuls vermells en l’any posterior al tractament. Els altres 3 pacients han vist reduïda la necessitat de transplantament en més d’un 70%.
Els efectes secundaris són els habituals d’un transplantament autòleg de moll de l’os: nàusees, fatiga, febre i major susceptibilitat a contraure infeccions.
Lligams:
- MHRA authorises world-first gene therapy that aims to cure sickle-cell disease and transfusion-dependent β-thalassemia, comunicat de premsa del 16 de novembre del 2023.
- L’edició genòmica per CRISPR: Charpentier i Doudna, Premi Nobel de Química 2020.
Cap comentari:
Publica un comentari a l'entrada